Our group develops and utilizes new light sources and techniques to follow the motions of electrons, holes, and nuclei in molecular and condensed matter systems on ultrafast time scales. Developing new technologies and physics ideas go hand in hand with gaining insight into ultrafast dynamics. We have built several novel instruments in the lab based upon our home-built frequency comb lasers. Some research themes appear below.
Frequency Combs
Frequency comb lasers, recognized with the 2005 Nobel prize in physics, have revolutionized atomic clocks and precision measurement. However, their enormous potential for ultrafast time-resolved measurements has been largely unexplored. The exquisite coherence of the pulse train of a frequency comb enables the signals from successive pulses to be coherently added and stored. This leads to orders of magnitude improvement in the attainable signal to noise of time-resolved experiments, enabling new and exciting directions that simultaneously push the boundaries of our ability to probe matter in the time and frequency domains. Now mature in the visible and near-infrared, recent breakthroughs have pushed frequency comb technology into more spectroscopically interesting regions of the electromagnetic spectrum. Mid-IR combs enable the probing and excitation of vibrational dynamics and UV combs enable the probing of electronic dynamics.
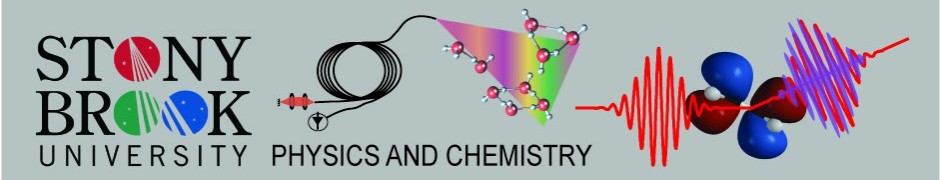
A frequency comb is formed by a train of well controlled femtosecond pulses.
Our frequency comb lasers have been based on Ytterbium (Yb) doped optical fibers. The small quantum defect, high doping, and the availability of double clad large-mode-area photonic crystal fibers makes Yb ideal for scaling to high average power. We have built in our lab an 80 W Yb:fiber laser for generating high order harmonics at high repetition rate. A detailed “how-to” paper is here.

High-power Yb-doped fiber amplifiers in the Allison lab
To make widely tunable frequency combs for cavity-enhanced optical spectroscopy (see below), we have also started branching out into using Erbium-doped fiber lasers frequency shifted in highly nonlinear fibers (HNLF). Using this backbone, we can make multiple shifted branches and then make widely-tunable mid-IR combs via difference frequency generation (DFG).

Left: A nonlinear Erbium-doped fiber amplifier, compactly fit into a small box. Right: Tunable frequency comb light spanning 1-2 microns. In this particular spectrum, the dispersive wave has been optimized for seeding our Yb-doped amplifiers.
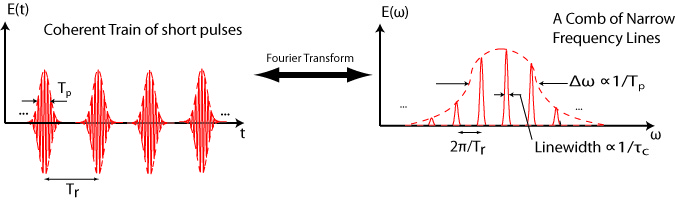
In total, using a combination of fiber lasers, HNLF broadening, optical parametric oscillators (OPO) and amplifiers (OPA), and high-harmonic generation, we have now in the Allison lab frequency combs spanning seven octaves of frequency space, as illustrated in the figure below.
A survey of frequency combs in the Allison lab, spanning seven octaves of frequency range. It has taken a lot of work to take combs out of their “comfort range” of the near-infrared, but it is the work necessary to move beyond demonstration experiments.
With new major funding from the NSF and the development of a new 2500 square foot lab space in the Physics building, we are expanding this range to cover from the THz to the soft x-ray, or 17 octaves (about 2.5 pianos worth) of frequency space.
Cavity-Enhanced High Harmonic Generation
High harmonic generation (HHG) can be used to generate ultrashort pulses of extreme ultraviolet (XUV) and soft x-ray light. These photon energies are ideal for performing photoelectron spectroscopy from surfaces and probing absorption edges of soft matter. However, due to limitations in laser power, most high-harmonic generation systems run at repetition rates around 1-10 kHz. We generate harmonics at repetition rates around 100 MHz, using the technique of cavity-enhanced high-order harmonic generation. In the cavity enhancement technique, a frequency comb laser is resonantly enhanced in an optical cavity whose repetition rate and carier-envelope offset frequency are matched to that of the laser. With kiloWatts of intracavity laser power, efficient HHG can be done at the full repetition rate of the mode-locked laser.
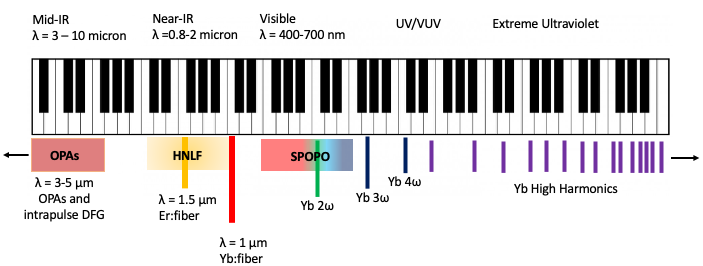
These sources are now among the world’s brightest XUV sources. In addition, the higher repetition-rate is particularly critical when recording photoelectron signals from surfaces, where space-charge puts severe limitations on the number of electrons per pulse that can be extracted from the sample. At synchrotron beamlines, nano-Ampere photocurrents parsed by sophisticated electron energy analyzers enable detailed studies of the electronic structure of solids and surfaces in energy, momentum, and spin. However, historically photoemission studies using XUV light produced from lasers have been limited to the pico-Ampere range, drastically limiting what can be studied. Using frequency comb methods, our system at Stony Brook can now produce space-charge-free photoemission currents also in the nA-range. For the purpose of surface photoemission, our system can be thought of as “table-top synchrotron” with ~1000 times better time resolution. A recent paper showing the performance of the source appears here.
A. Schematic of the Stony Brook 61 MHz tr-ARPES machine. B. Photograph of the setup. C. Example time-of-flight ARPES data. D. Illustration of parallel full 3D data collection of both un-excited electrons (orange) and excited electrons (blue).
Time- and Angle-Resolved Photoemission
An electron’s momentum parallel to the surface is conserved in the photoemission process. This allows photoemission to report on both the energy and momenta of electrons in solids, and angle-resolved photoelectron spectroscopy (ARPES) using synchrotron radiation has become and indispensable tool for determining the electronic structure of matter. ARPES can be used for determining the band structure of solids, the electronic structure of more complicated “quantum materials” with strong electron correlation, and also reconstruct orbital shapes for molecules on surfaces.
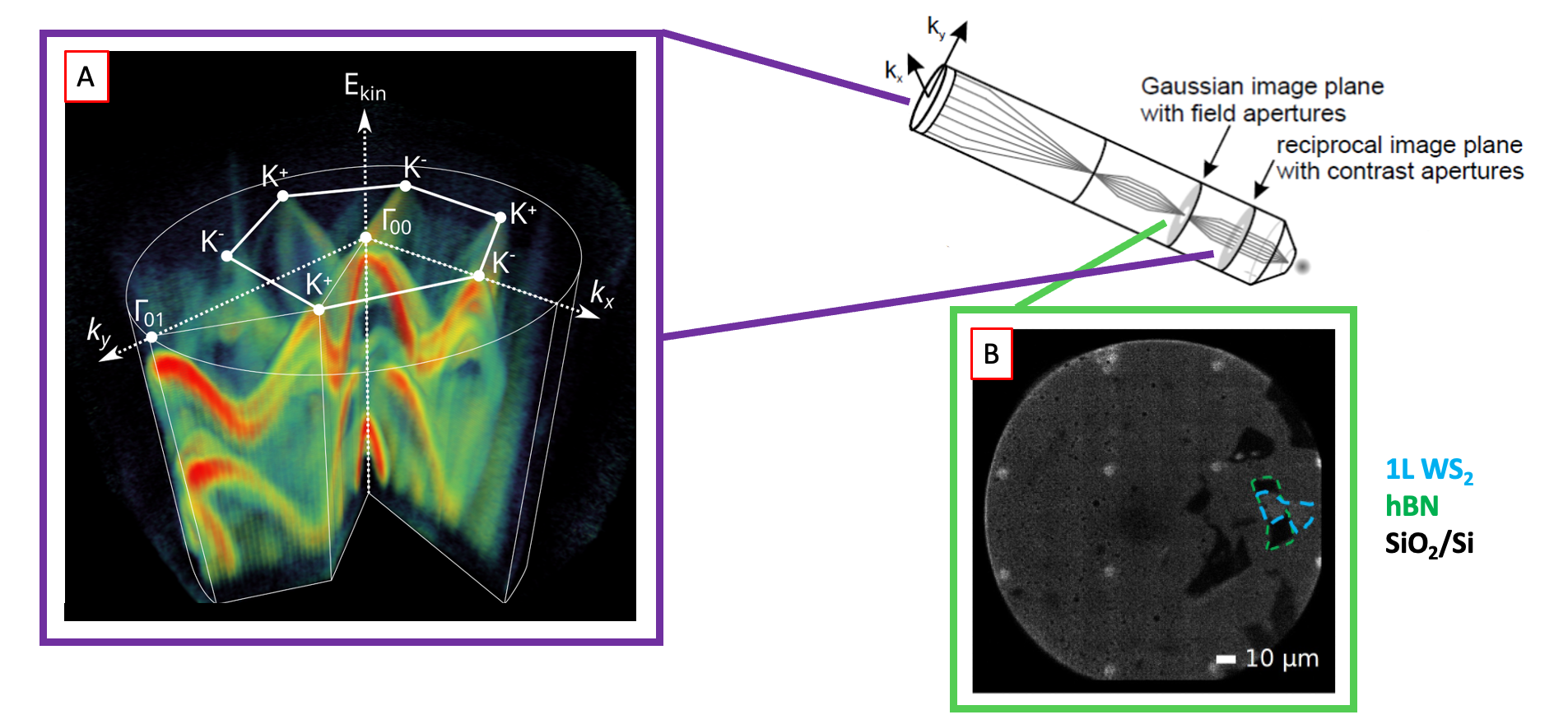
A. Full valence band structure for monolayer tungsten disulfide (WS2) measured from a 10 micron exfoliated flake in seconds using the parallel detection of the momentum microscope. B. Real-space PEEM image of the sample.
With support from the Stony Brook Foundation’s Discovery Prize, and in collaboration with the Schönhense group in Mainz, we have implemented time-of-flight momentum microscopy on our ARPES endstation. This new analyzer collects all emitted photoelectrons in parallel and gives an improvement of several orders of magnitude in the data rate for ARPES measurements, enabling the study of perturbatively excited samples. The momentum microscope also enables selection of micron-scale spatial region on the sample, for time-resolved micro-ARPES studies.
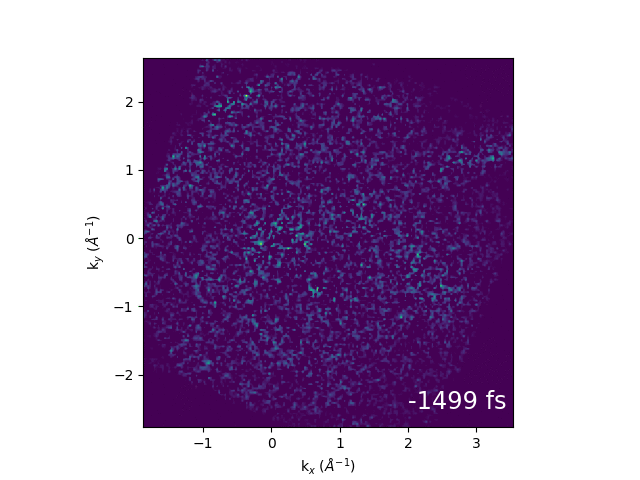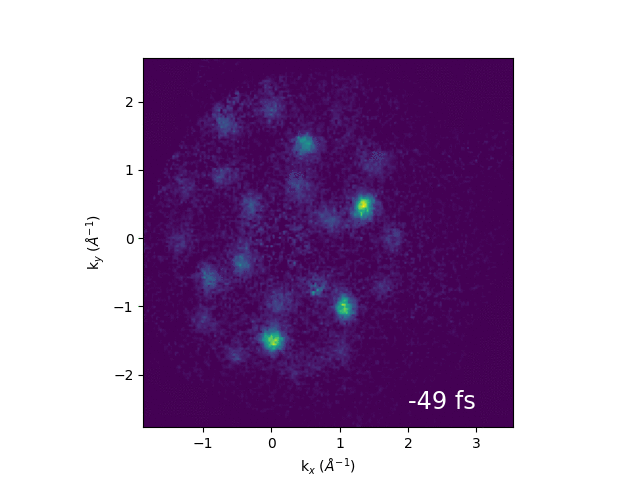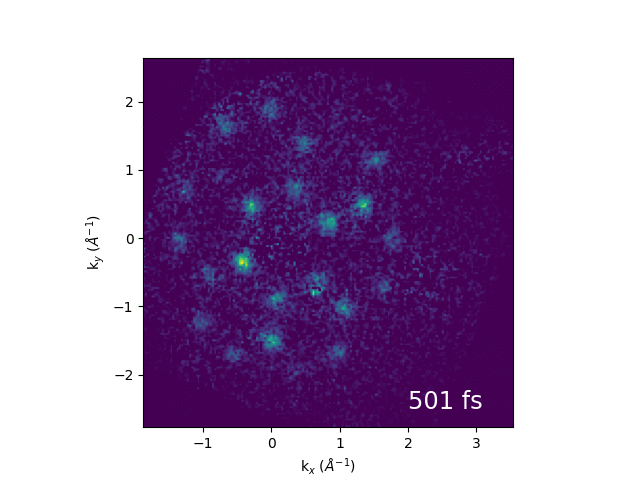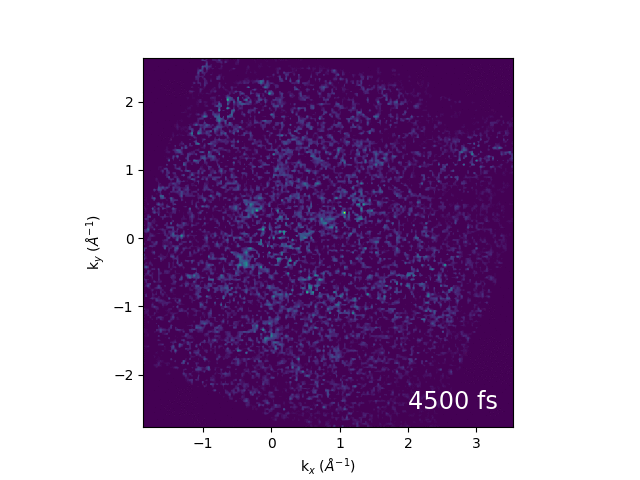
An example of electron dynamics in the bands of bulk WS2 is shown below. Our first publication from the combination of momentum microscopy and our 61 MHz cavity-enhanced HHG source can be found here.
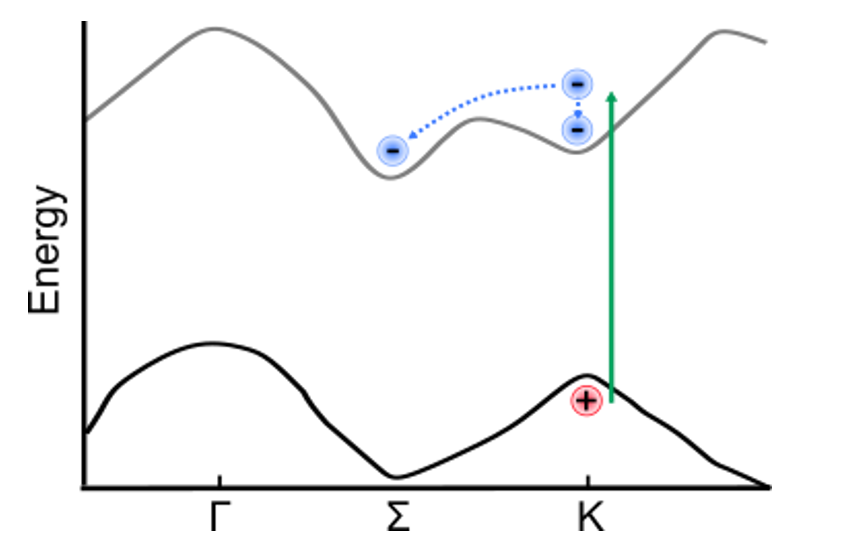
Illustration of band structure of WS2. Electrons are excited by 517 nm light across the direct band gap at the K point of the Brillouin zone, then relax to conduction band minimum. The GIF to the right shows momentum-space images of the dynamics.
Cavity-Enhanced Ultrafast Spectroscopy
Ultrafast spectroscopy is widely applied to a range of systems from molecules in solution to isolated molecules in the gas phase. However, measurements in the gas phase are almost always done using photoionization, due to the very high sensitivity of charged particle detection, whereas measurements in solution are restricted to measuring absorbed or nonlinear scattered light. Photoionization of gas phase clusters is also problematic due to cluster fragmentation. Comparison of gas phase, cluster, and solution phase data is thus often highly nontrivial.
We have developed techniques using frequency combs and optical resonators to achieve a large sensitivity improvement in ultrafast spectroscopy, so that optical signals can be recorded from dilute molecular beams. We have demonstrated transient absorption spectroscopy with detection limits as low as ΔOD = 2×10¯¹º, and also published schemes for using multiple combs for recording cavity-enhanced multi-dimensional spectroscopy signals. This allows the direct comparison of solution-phase and gas-phase data, and allows ultrafast vibrational spectroscopy to be applied to gas-phase and cluster systems.
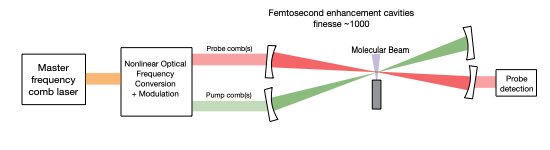
The basic principle of cavity-enhanced ultrafast spectroscopy. Pump and probe combs are both resonantly enhanced in optical cavities. The nonlinear spectroscopy signal is enhanced by a factor proportional to the cavity finesse squared, enabling large signal enhancements even for cavities of modest finesse.
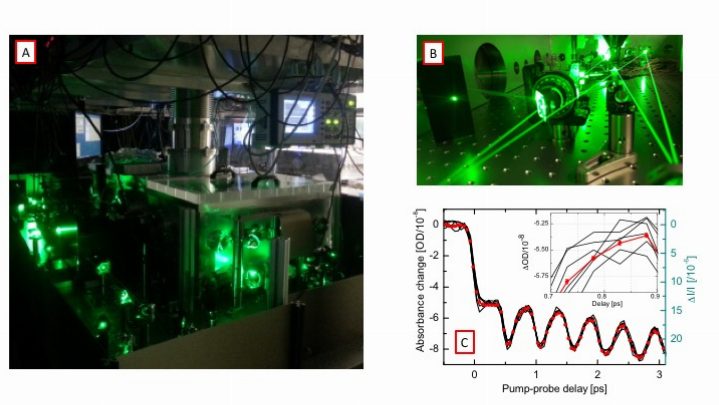
A and B: The cavity-enhanced ultrafast spectrometer. Two femtosecond enhancement cavities are built on a vibration isolated platform inside the vacuum chamber. C: Transient absorption data recorded form molecular iodine with a noise level of ΔOD = 2E-10.
After initial demonstration experiments in 2016, substantial work has gone into developing widely tunable versions of these spectrometers operating in the UV, visible, and infrared. This has required breaking new ground both on frequency comb generation and cavity enhancement. An example showing widely tunable cavity-enhanced frequency combs is below. We have now achieved comparable noise performance with this tunable spectrometer as our one-color demonstration and have been doing detailed studies on molecules undergoing excited state proton transfer, internal conversion, and intersystem crossing. A schematic of the broadband CE-TAS spectrometer is shown below as well as transient absorption spectra of jet-cooled salicylideneaniline.
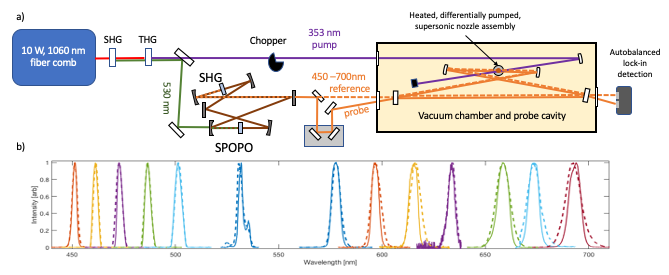
a) The tunable UV/VIS CE-TAS Spectrometer. Tunable combs from a synchronously pumped optical parametric oscillator (SPOPO) are resonantly enhanced in a 4-mirror dispersion-managed ring cavity. b) Intracavity spectra (solid) and spectra incident on the cavity (dashed). The intracavity GDD is low enough that bandwidth reduction due to the cavity is minimal.
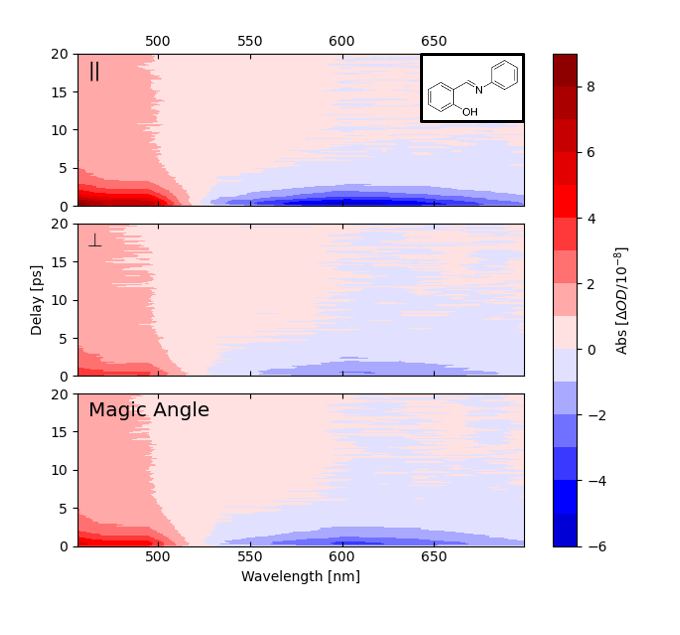
Transient absorption spectra of jet-cooled salicylideneaniline with pump and probe polarization parallel (top), perpendicular (middle), and at the “magic angle” of 54.7 degrees. Pump wavelength is 353 nm. The molecules are at a temperature of approximately 40 K, estimated from the decay of the rotational anisotropy.
Future work is planned for the development of cavity-enhanced ultrafast spectrometers operating in the infrared for recording 2DIR spectra of dilute gasses. This has applications in both basic science (e.g. ultrafast 2DIR of hydrogen-bonded clusters) as well as major real-world applications in trace-gas analysis.